Log in to your home directory on the XSEDE VM and copy the files to your home directory:
cp -r moledata/modsel_sim_tutorial/ .
cd modsel_sim_tutorial/
jModelTest GitHub repository: https://github.com/ddarriba/jmodeltest2
For most of this tutorial we’ll be using PAUP*, but we’ll do one simple example with jModelTest here; refer to the manual at the link above for details on how to use the program.
We’ll use the primate-mtdna.nex data file.
We will evaluate 56 models on a tree obtained using BioNJ with Jukes-Cantor distances. We’ll use both the corrected AIC (AICc) and the Bayesian Information Criterion (BIC):
jmodeltest -d primate-mtdna.nex -s 7 -t fixed -g 4 -f -i -AICc -BIC -tr 2
(Pause while we discuss the output)
We will use the command-line version of PAUP* installed on the server. GUI versions of PAUP* are also available for macOS and Windows. If you want to install them on your laptop, you can download them from this page: http://phylosolutions.com/paup-test/
Load the primate-mtdna.nex file into your favorite editor (e.g., nano, vi, or emacs). After I have made some comments, exit the editor without saving any changes.
For the first run, we will type PAUP* input commands interactively. Start the program and execute the primate-mtdna.nex data file:
paup primate-mtdna.nex -L log.txt
The -L switch above opens a log file that will receive all output that is displayed on the screen during the run.
Now, we will attempt to exactly reproduce the jModelTest analysis. First, we need a tree, so we’ll compute a BioNJ tree using Jukes-Cantor distances:
dset distance=jc;
nj bionj=y;
We’ll use the automodel command to perform model selection. First, let’s see what the available options and current settings are:
automodel ?
The current settings look fine, so let’s start the run:
automodel;
The AICc criterion chooses the Tamura-Nei+Gamma (TrN+G) model, and this model is automatically set up in PAUP* for subsequent analysis, using the fixed parameter values just estimated. You can compare the output from PAUP* to the previously obtained jModelTest results.
We can now search for a maximum likelihood tree in PAUP*. First, we have to change the active optimality criterion to maximum likelihood (the default criterion is parsimony, not because I think parsimony is a good method, but because it can be used on any kind of character data).
set criterion=likelihood;
Remember that you can always abbreviate commands and options as long as they remain unambiguous, so the above command could also have been entered as:
set crit=l;
IMPORTANT: Searches in PAUP are extremely slow if model parameters are estimated during a tree search. It is almost always better to estimate model parameters on a fixed tree, and then fix those parameters prior to initiating the tree search.* The “automodel” command does this for you. However, if you estimate model parameters manually, you need to run the command lset fixall; to fix the model parameters to the estimated values.
Initiate a heuristic search using the default search settings:
hsearch;
Examine the tree that was found:
describe/plot=phylogram;
A somewhat safer searching strategy is to use different starting points, similar to RAxML, Garli, and other programs. The usual way to this in PAUP* is to use “random addition sequences” to generate different starting trees by stepwise addition. For difficult data sets, many starting trees for branch swapping will be different, and the searches may land in different parts of “tree space.” If all random-addition sequence starts end up finding the same tree, you can be reasonably confident that you have found the optimal tree(s).
For the sake of completeness, let’s do a random-addition-sequence search using the current model settings:
hsearch addseq=random nreps=50;
Obviously, this is an “easy” data set: most, if not all, starting points end up finding the same tree.
PAUP* now has the ability to initiate searches using RAxML, Garli, and FastTree2, (and I plan to add PhyML, etc., as well). The current PAUP* model settings are used to determine the option settings for these programs. Input and/or config files are created, the program is invoked as an external command, and the resulting trees are imported back into PAUP*. For example, let’s look at the options available for the raxml command in PAUP*:
raxml ?
(I will make a few comments here.)
Start a RAxML search after changing the ML settings to optimize all parameters:
lset estall;
raxml;
Now let’s optimize the tree from RAxML in PAUP*:
lscores;
Note that the likelihood score from PAUP* is slightly worse than the one reported by RAxML. This is because PAUP* is using the Tamura-Nei model rather than the more general GTR model, which RAxML does not support. To validly compare likelihoods, we need to set up the model in PAUP* to match the one used by RAxML. This involves undoing the restriction on the GTR substitution rates imposed by Tamura-Nei:
lset rclass=(abcdef);
lscores;
The previous (Tamura-Nei) model had set rclass=(abaaca), so that all 4 of the transversions are occurring at the same rate, but each of the transitions are potentially occurring at distinct rates. After the change to ‘rclass’ PAUP* gets a slightly better likelihood than RAxML. Presumably, PAUP* just did a slightly better job at optimizing the model parameters and branch lengths (I have worked very hard on this aspect of the program).
PAUP* can do “partitioned” analyses in which different models are assigned to different subsets of sites (e.g., genes, or codon positions, or a combination of these). The easiest way to set up a partitioned analysis is to use the autopartition command.
Partitioning begins by defining a set of basic “blocks”, which are the smallest divisions of sites. The goal is to see if an adequate model can be achieved by combining some of these blocks into larger ones, thereby reducing model unnecessary model complexity (and speeding searches because fewer parameters need to be estimated).
The primate-mtdna.nex data file defines a partition called codons that places sites into one of four categories: 1st, 2md, and 3rd positions, plus non-protein-coding (tRNA) sites. Let’s first look at the options:
autopartition ?
Now start an automated partitioned analysis (this may take a few minutes on a heavily loaded machine):
autopartition partition=codons;
Partitioning by BIC combines 1st position and noncoding blocks into the same larger subset.
You can evaluate the likelihood of the resulting model with the lscores command:
lscores;
Before starting a search, you should fix all model parameters to the MLEs obtained above rather than optimizing parameters during the search (which is exceedingly slow!)
lset fixall;
lscores;
The second lscores command above verifies that the parameters have been fixed to the MLE values. Now you can do a search as before:
set crit=like;
hsearch;
describe/plot=p;
Note that PAUP*‘s autopartition capability was inspired by Rob Lanfear’s PartitionFinder program (http://www.robertlanfear.com/partitionfinder/), which has a few extra features. There is a nice online tutorial at http://www.robertlanfear.com/partitionfinder/tutorial/.
The above is intended to just give a flavor of how PAUP* works. We’ve barely scratched the surface with respect to what it can do. PAUP* can perform parsimony analysis under a variety of cost schemes, neighbor-joining and criterion-based distance searches using a large number of distance transformations, exact tree searches for smaller numbers of taxa, consensus tree calculation, agreement subtrees, distances between trees, analysis of base frequency composition, likelihood under amino-acid models, likelihood for discrete character data, and much more.
If you have time, you might also like to go through the tutorial I have used in previous versions of the course: https://molevol.mbl.edu/index.php/PAUP*_Exercise. This tutorial demonstrates additional capabilities and features of PAUP*, and provides more background on tree-searching, model selection, bootstrapping, etc.
But now it’s time to move on to simulations…
Uses pseudorandom numbers to draw:
Simulating sequences on a tree:
We’ll use PAUP* as our simulator for this exercise. There are other good simulators out there, notably:
seq-gen (Andrew Rambaut): http://tree.bio.ed.ac.uk/software/seqgen/
The nice thing about PAUP*‘s simulation capability is that you don’t have to write pipelines to pass simulated data to inference programs – it’s all internal!
We’ll start with a very simple 3-sequence simulation (sim-3taxa.nex):
#nexus
[ This example just simulates three sequences under the HKY model ]
begin paup;
cd *;
set warntree=no notifybeep=no;
end;
begin taxa;
dimensions ntax=3;
taxlabels X Y Z;
end;
begin trees;
tree 1 = [&R] ((X:0.05,Y:0.05):0.45,Z:0.5);
end;
begin dnasim;
simdata nchar=1000;
lset nst=6 basefreq=(.1 .2 .3 .4) rmatrix=(1 4 1 1 4 1);
truetree source=memory treenum=1 showtruetree=brlens;
beginsim nreps=1 seed=0 monitor=y;
[ Ordinarily, there will be commands in here, but we're just
getting started. You don't have to include this comment,
but if you do don't forget to close the opening square
bracket with a closing square bracket! ]
endsim;
end;
To avoid possible problems with settings left over from the previous analyses, let’s restore PAUP’s internal state to the settings in effect when it first launches:
reset factory;
Now execute the file:
execute sim-3taxa.nex;
Look at your simulated data by typing the command:
showmatrix;
How can we get a sense of how well the simulated sequences correspond to our “true” tree and model? One thing we can do is calculate model-based evolutionary distances (sometimes called “corrected” distances). First, show the distances calculated without model-based correction:
dset distance=p;
showdist;
Do the distances correspond to the expected number of substitutions along the tree? Are they underestimates or over-estimates of the number of substitutions? Is the degree of underestimation related to the level of divergence implied on the true tree?
Now let’s try some model-based distance transformations. Let’s use the simplest model that attempts to account for unobserved substitutions, the Jukes-Cantor model.
dset distance=jc;
showdist;
The distances are closer to the expected values, but the larger distances are still substantially underestimated. Next, we can into account the bias in transition/transversion rates using the Kimura 2-parameter distance:
dset distance=k2p;
showdist;
The distances are closer to the expected values, but the larger values are still too low. This is because we are still assuming equal equilibrium base frequencies, but the true frequencies are unequal. We can deal with the unequal frequencies (but not the transition bias) using the Felsenstein 1981 (F81) model.
dset distance=f81;
showdist;
The Hasegawa-Kishino-Yano HKY) model handles both the transition bias and unequal base frequencies:
dset distance=hky;
showdist;
Now the distances correspond closely to the expected values (and would be even closer if longer sequences were used).
Finally, let’s use a model that is more complex than the true process responsible for generating the data, the General Time Reversible (GTR) model:
dset distance=gtr;
showdist;
As you can see, the calculated distances are still very accurate (the model is correct, just more general than necessary). However, the variance of the evolutionary distances estimates will be slightly higher in this case.
Now we will run a more complicated simulation that demonstrates the problem of long-branch attraction. I’ll keep it simple here, but you can modify the example if you like to see what the effect of other kinds of model misspecification might be, etc.
Here is the file (sim-4taxa.nex)
#nexus
[ This example demonstrates the dreaded Felsenstein zone. ]
begin paup;
cd *;
set storebrlens nostatus autoclose=yes warntree=no notifybeep=no;
end;
begin taxa;
dimensions ntax=4;
taxlabels A B C D;
end;
begin trees;
tree 1 = [&R] ((A:1.0,B:0.1):0.1,(C:0.1,D:1.0):0.1);
end;
begin dnasim;
simdata nchar=(10 100 1000 10000);
lset model=jc nst=1 basefreq=eq;
sitemodels jc:1;
truetree source=memory treenum=1 showtruetree=brlens;
beginsim nreps=100 seed=0 monitor=y resultsfile=(name=sim4.results replace output=means);
[parsimony]
set criterion=parsimony;
alltrees;
tally parsimony;
[likelihood under JC]
set criterion=likelihood;
lset basefreq=equal nst=1;
alltrees;
tally 'ML-JC';
[NJ using P distances ]
set criterion=distance;
dset distance=p;
nj;
tally 'NJ-P';
[NJ using JC distances]
set criterion=distance;
dset distance=jc;
nj;
tally 'NJ-JC';
endsim;
set monitor=y;
end;
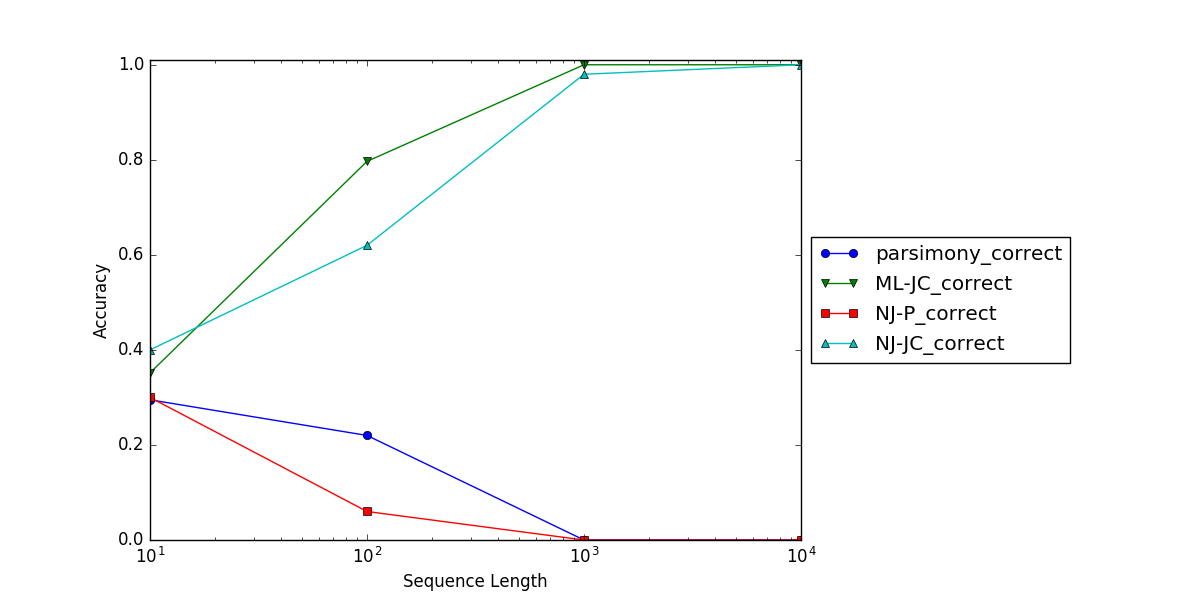
The tree is a four taxon tree with two long branches and three short branches. The “nchar” option on the simdata command specifies the number of sites simulated in each replicate (we’ll simulate four different sequence lengths). The substitution model is Jukes-Cantor. For each simulated sequence length we’ll analyze the results using 4 methods: parsimony, likelihood (assuming the correct model), neighbor joining (using uncorrected P distances), and neighbor joining using Jukes-Cantor distances. We’ll do all of this 100 times.
After the simulation completes, the sim4.results file will contain the output of the “tally” commands which keep track of how often the correct tree was obtained. The results of one run of this simulation are shown in the figure below.
In our last exercise, we will simulate a data set under the Tamura-Nei model with gamma-distributed rates across sites, and evaluate the performance of various model selection criteria.
First, execute a file which we’ll use to simulate one data matrix (sim-6taxa.nex).
execute sim-6taxa.nex;
Now get a starting tree and run an automated model selection:
dset distance=logdet;
nj;
automodel;
After we discuss the results, let’s also do a likelihood ratio test (LRT). We will compare three models: the Jukes-Cantor model, the true model (TrN+G), and the most complex nucleotide model available in PAUP* (GTR+I+G). I will show you how to set up and run these models. We’ll then do LRTs manually and compare our results (everyone will have simulated a different data set). You can use the calculator here to calculate P-values for the LRT.