MUSCLE Background
Muscle is described in Edgar et al. 2004.
Muscle is a modified progressive alignment algorithm which has comparable accuracy to MAFFT, but faster performance. Muscle has remained popular for phylogenomic studies, and there has been some evidence that Muscle is more accurate than MAFFT on amino acid alignments (Pais et al. 2014).
Broadly it consists of three stages:
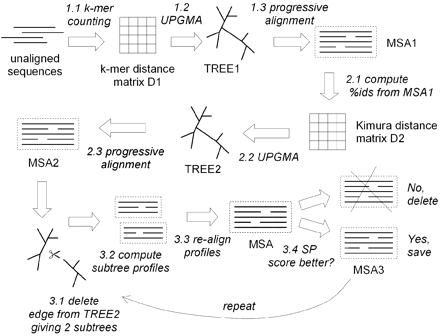
Stage 1. Draft progressive alignment. The goal of the first stage is to produce a multiple alignment, emphasizing speed over accuracy, using an approximate kmer distance measure. Kmer distance between two sequences is defined by first collecting the set of k-mers (subsequences of length k) occuring in the two sequences, then measuring how different the two sets are.
Stage 2. Improved progressive. The main source of error in the draft progressive stage is the approximate kmer distance measure, which results in a suboptimal tree. MUSCLE therefore re‐estimates the tree using the Kimura distance, which is more accurate but requires an alignment.
Stage 3. Refinement. The tree is broken into subtrees, and the sub-alignments refined.
References
Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792-1797.
Pais FS-M, de Cassia Ruy P, Oliveira G, Coimbra RS. 2014. Assessing the efficiency of multiple sequence alignment programs. Algorithms Mol Biol. 9:4.